Molecular Dynamics (MD) simulations offer the opportunity to dynamically study complex biological and material processes at temporal and spatial resolutions beyond the scope of experimental methods. Among the biological processes, signalling pathways, enzyme regulation, protein folding and interactions, membrane composition, and transmembrane transport are of particular interest, as they are key to understanding the development, progression, and treatment of severe diseases such as cancer and neurodegenerative disorders. In our group, we are working on a wide range of corresponding research topics, including the simulation of receptor and ligands that are important for apoptosis and signalling processes, parametrization of force field parameters for post-translational modifications and metal organic frameworks, and the development of a machine learning framework for resolution transformation. Specific projects can be found here.
Research Highlights
Vastly different energy landscapes of the membrane insertions of monomeric gasdermin D and A3
Membrane pores allow the transport of molecules across lipid bilayers. They play a key role in biological mechanisms, among them pyroptosis, a form of programmed cell death. One class of pyroptotic agents are gasdermins, a family of pore forming proteins which are activated as part of immune responses, but have also been linked to various diseases. Gasdermins share the same secondary structure, though their amino acid sequences differ, which is being reflected in their behavior. Our latest molecular dynamics simulations (see our original research paper for more details) show that gasdermin D and A3 follow distinct paths of pore formation, also depending on the membrane composition: In case of a native-like E. coli bilayer, gasdermin A3 energetically prefers the membrane-adsorbed state over the inserted one, while gasdermin D, thanks to numerous charged and polar residues on the pore-lining β-hairpins interacting with lipid headgroups (especially phosphatidylethanolamine) and water molecules, meets a much smaller insertion barrier and settles in a deep free energy minimum inside the bilayer. On the other hand, if the membrane lacks certain lipids, a behavior analogous to gasdermin A3 is observed.

Molecular details of novel apoptotic pathway
Apoptosis is controlled by interactions of pro- and anti-apoptotic proteins from the Bcl-2 family of proteins. An excess of pro-apoptotic proteins results in the formation of pore-forming oligomers capable of disrupting the mitochondrial outer membrane, a process ultimately leading to cell death.
Here in collaboration led by Tobias B. Beigl and Frank Essmann at the Robert Bosch Center for tumor diseases on the Bosch Health campus, our master's student Thomas Fellmeth, currently doing his PhD in CEITEC in Brno, Czech Republic, studied the oligomerization of transmembrane anchors of pro-apoptotic BOK and anti-apoptotic BCL-2 proteins in endoplasmic reticulum. Together the extensive experimental and simulation work has revealed that (i) the peculiar BOK protein is indeed causing cell death, (ii) this deadly effect can be mitigated by the BCL-2 protein, (iii) the interaction between the two proteins is mediated by their transmembrane anchors, and (iv) BOK and BCL-2 are thereby located in the endoplasmic reticulum instead of the traditional mitochondrial outer membrane. Moreover, Thomas's molecular dynamics simulation predicted BCL-2 residues essential for interaction with BOK that have been confirmed by Tobias experimentally. To sum up, our work published in the renowned open-access journal EMBO reports introduces a novel apoptotic pathway, and the interactions we have identified at the atomic level offer promising new targets for drug development.

Entropic barrier of water permeation through single-file channels
Splitting of Gibbs free energy of water permeation through single-file channels into its enthalpic and entropic contributions provides novel insight into the nature of the studied processes and may open new avenues for design of enthalpically vs entropically driven devices. In our recent paper, the combination of transition state theory with the transmission coefficient obtained from molecular dynamics and with high-resolution experimental measurements of water permeation through aquaporin-1 (AQP1) over a broad range of temperatures unravels that the total Gibbs free energy barrier of water permeation through AQP1 at 298K amounting 3.02 kcal/mol results mainly from the enthalpic barrier, being 3.16 kcal/mol. Despite orientational restriction of water molecules in the pore the barrier is slightly decreased by the entropy.
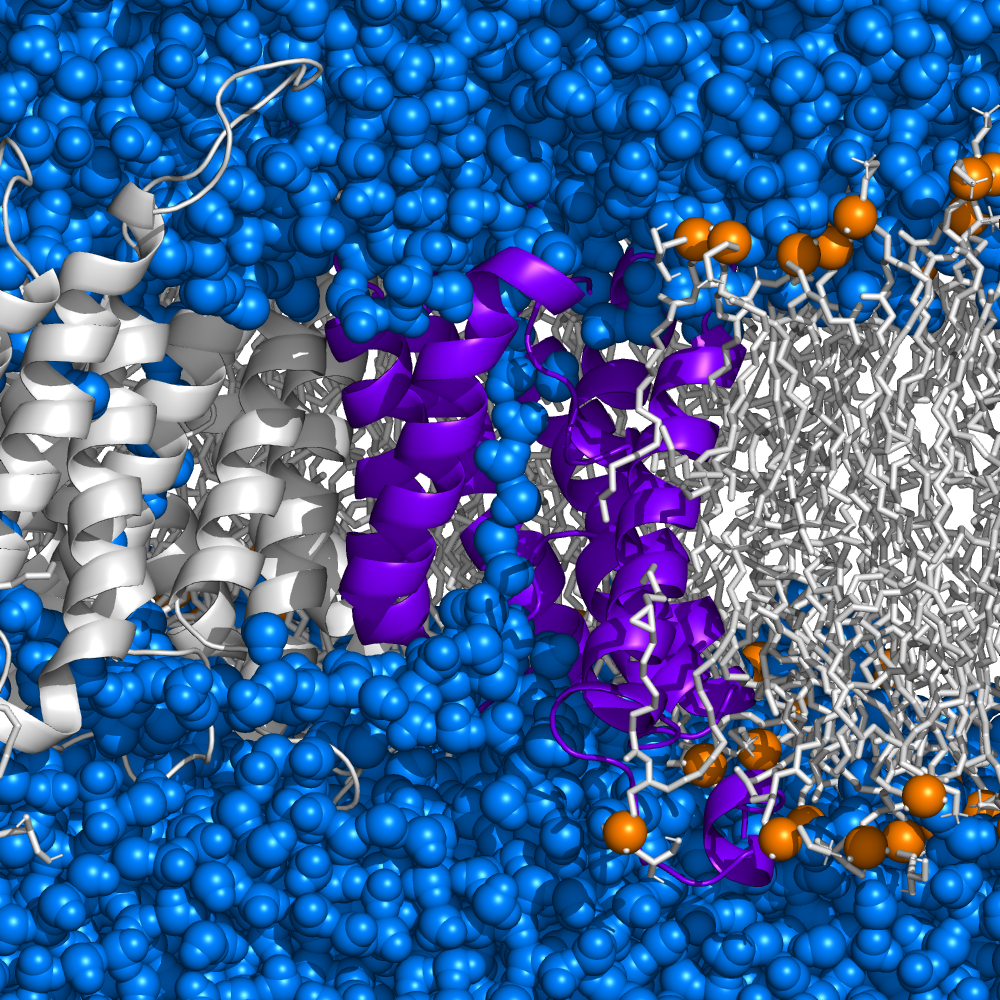
Ninjurin-1 Protein - Charon of cell life
Cell death is an essential process in our body that prevents the development of tumors and the spread of pathogens in the body through the controlled elimination of damaged or infected cells. Contrary to popular belief that cells simply burst at the end of their lives, a specific protein called ninjurin-1 was recently discovered to serve as a predetermined breaking point for cell membrane rupture. Now, together with an international team of researchers from the University of Basel, the University of Lausanne, and ETH Zurich we have elucidated the exact mechanism of the final step of cell death at the atomic level. In the groundbreaking publication in the journal "Nature", we describe how ninjurin-1 assembles into filaments that rupture the plasma membrane by stabilizing membrane edges.
Not a scientist and nevertheless interested in this topic? Then check the Labour Journal Article written by Anna Sternberg summarizing our work.
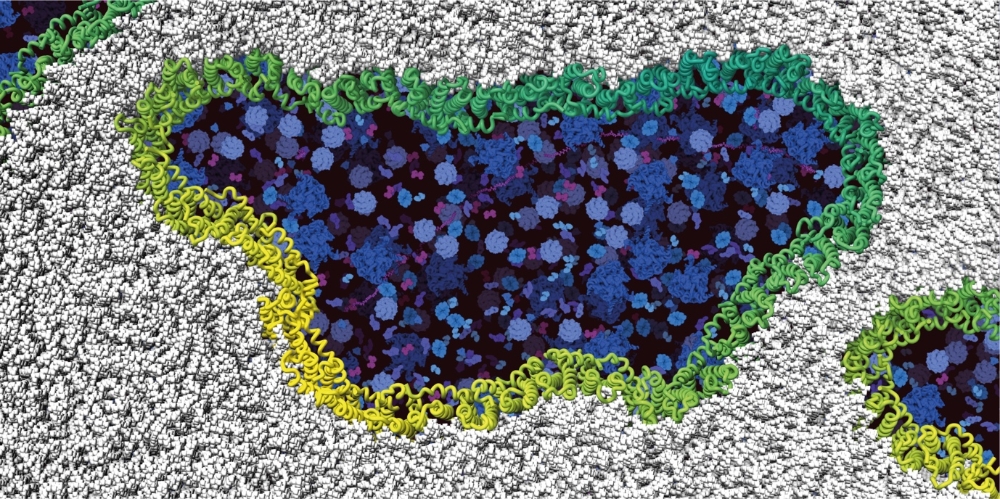
Complex regulation of water permeability through an aquaglyceroporin
In our recent joint experimental (group of Andreas Horner, JKU Linz, Austria)/simulational publication we unravel that, contrary to the common notion that aquaporins posses constitutively open pores, multiple pore lining residues are responsible for modulation of water permeability through the aquaglyceroporin GlpF. The impact of each individual position was only able to be quantified using Bayesian generalized multilevel models in collaboration with Paul-Christian Bürkner (SC SimTech). Moreover, we have successfully designed and characterized mutants closing or opening the water pore.
Mysterious sorcery unmasked! How charged residues influence the membrane insertion of gasdermin-A3
In our publication in Frontiers in Cell and Developmental Biology we unveil the mystery behind the ability of gasdermin's spontaneous membrane insertion and thus the initiation of the gasdermin pore formation. Our atomistic MD simulations of monomeric gasdermin-A3 (GSDMA3) and small arcs inserted in an E. coli polar lipid extract membrane reveal the astounding effect that salt-bride formation and protein surroundings have on the energetics of GSDMA3's transmembrane passage. Furthermore, our simulations support the hypothesis of oligomers preassembling on the membrane surface prior to membrane insertion, though the oligomer can be significantly smaller than a full pre-pore rings.

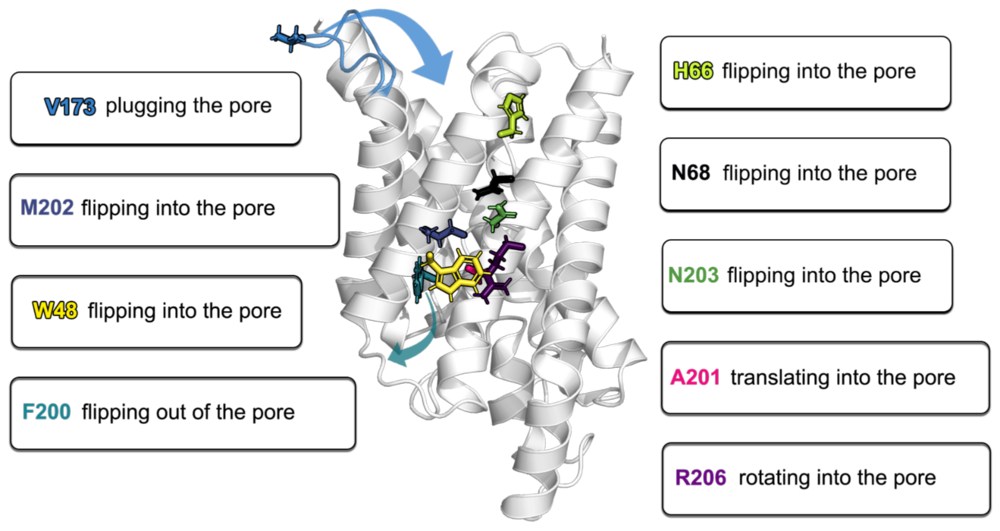
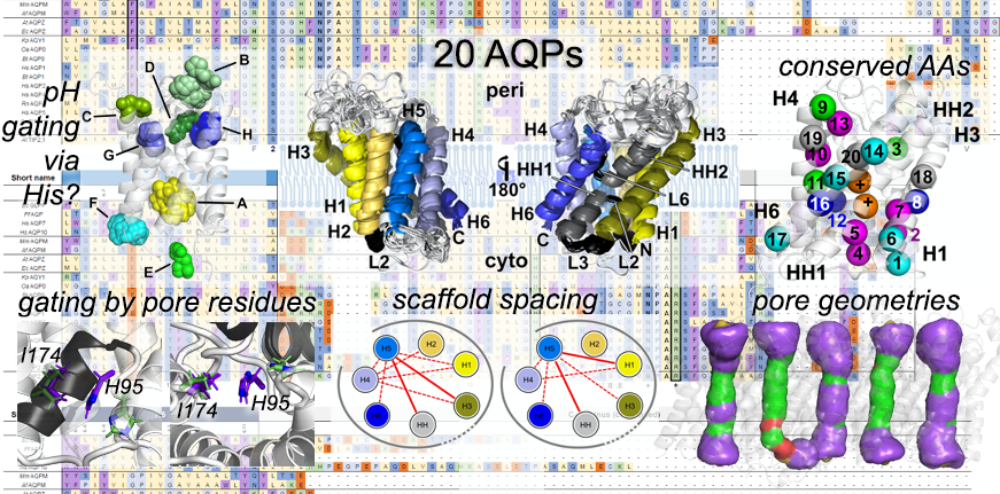
The hidden intricacies of aquaporins
Our recent publication in Small uncovers remarkable details and (dis)similarities in the common structural scaffold of aqua(glycero)porins. Thereby, MD simulations have bestowed the investigated X-ray structures the dynamics neccesarry for understanding of e.g. gating behavior by pore lining residues.

Cholesterol stabilizes GPCRs only at physiological temperature
Cholesterol is known to influence many functions in our bodies. Our recent publication in Science Signalling reveals by combination of atomic force microscopy with molecular dynamics simulations how cholesterol analog stabilizes a prominent G Protein-coupled receptor, β2-adrenergic receptor, specifically at body temperature of 37°C. The cover image depicts the receptor interacting with the cholesterol analog, shown as green surfaces, in liposomes at 37℃.
Proteolytic pore formation in lipid membranes by Gasdermin-A3
Gasdermins are main effectors of pyroptosis, an inflammatory form of cell death. In our recent publication, the combination of state-of-the-art atomic force microscopy and extensive multiscaling molecular dynamics simulations sheds light on (i) assembly of gasdermin-A3 oligomers on membrane surface, (ii) growth and reshaping of membrane-inserted gasdermin-A3 oligomers and (iii) pore formation by lipid unplugging. Related publication: Mari & Pluhackova et al. Nature Communications 2022
How YidC insertase facilitates membrane insertion
If you are interested in how our multiscaling molecular dynamics simulations revealed parts of the membrane insertase YidC responsible for membrane insertion of the protein Pf3 check our recent publication in Nature Communications. Our simulations complement a wealth of experimental investigations (performed at the ETH Zürich and at the University of Hohenheim) that altogether reveal that the initial contact between the insertase and the substrate is formed by the YidC's cytoplasmic α-helical hairpin domain. However, it is the interaction of the substrate with the hydrophilic groove of YidC which decides about the membrane insertion and its efficiency.
{kind=link}