Biomarker sind biologische Merkmale, die im Blut oder im Gewebe nachgewiesen werden können. Das können beispielsweise Genveränderungen sein, wie sie in Krebszellen vorkommen. Biomarker können deshalb Auskunft geben über spezielle Eigenschaften eines Tumors. Anhand von Biomarkern kann auch eine individuelle Therapie ausgewählt werden.
Brustkrebs ist immer noch die zweithäufigste Krebstodesursache bei Frauen, trotz beträchtlicher Fortschritte bei Prävention, Diagnose und Patientenmanagement. Das Brustkrebsrisiko und der Krankheitsverlauf kann mit bestimmten Methoden prognostiziert werden, zum Beispiel anhand von einzelnen Biomarkern oder sogenannten Multi-Gen-Expressionstests. Letztere sind jedoch nur für Patientinnen zugelassen, die zu bestimmten Risikogruppen gehören.
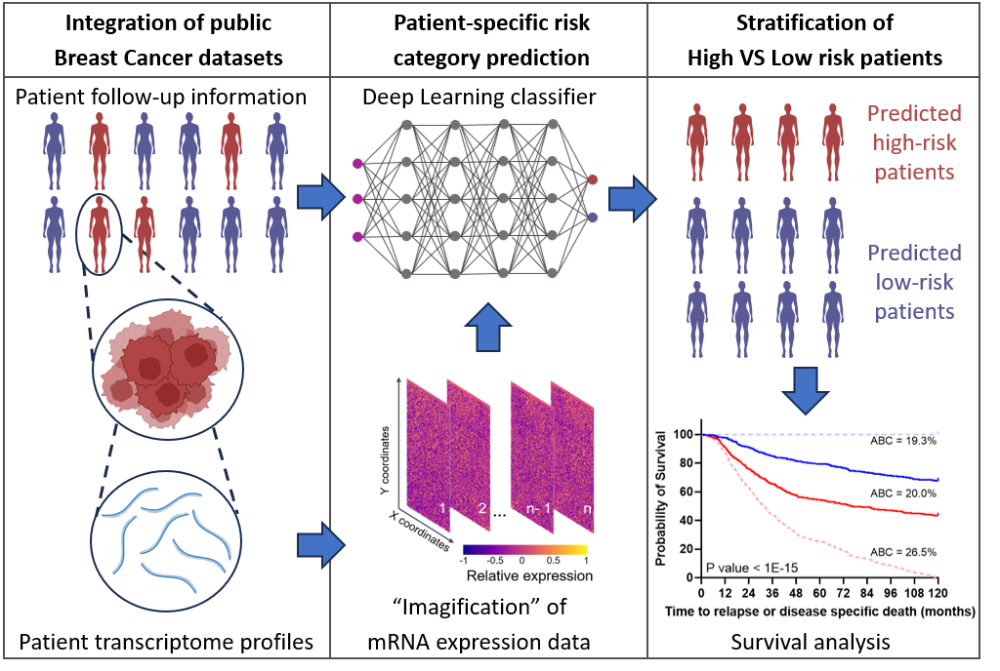
Deshalb haben Markus Morrison, Professor und Leiter des Instituts für Zellbiologie und Immunologie an der Universität Stuttgart und sein wissenschaftlicher Mitarbeiter Cristiano Guttà, in Zusammenarbeit mit der Ludwigsburger Firma ProKanDo, die auf IT-Security und KI-Lösungen spezialisiert ist, eine Methode entwickelt, die das Risiko für alle Patientinnen nicht nur anhand einzelner Biomarker oder mehrerer Hochrisikogene ermittelt, sondern das komplette genetische Expressionsprofil eines Menschen berücksichtigt. Das sind rund 18 000 Gene pro Person, die in die Simulation mit einfließen. Zusätzlich haben sie künstliche Datensätze generiert, um die Allgemeingültigkeit und Zuverlässigkeit der Prognosen zu verbessern.
Virtuelle Patientinnen verbessern die statistische Aussagekraft
Als Grundlage verwendeten die beiden Forscher zwei große Datensätze von Brustkrebspatientinnen, die sowohl Genexpressions-Informationen als auch klinische Daten enthielten, zum Beispiel, wie lange eine Patientin nach der Diagnose noch lebt.
Mit Multi-Gen-Expressionstests können mehrere Gene gleichzeitig analysiert werden. Mit den Tests werden komplexe biologische Zusammenhänge charakterisiert, Krankheiten diagnostiziert, Verläufe prognostiziert oder die Wirksamkeit von Therapien vorhergesagt.
Für statistische Vorhersagen ist es jedoch schwierig, durch die Vielzahl dieser 18 000 Gene pro Person und der bestehenden, im Vergleich relativ kleinen Gruppe mit rund 1 400 Patientinnen, gute Prognosen für die Allgemeinheit abzuleiten. Deshalb haben sie mit Hilfe einer künstlichen Intelligenz die Datensätze künstlich vergrößert. „Die Muster, die in diesen Patientendaten verborgen sind und mit dem Krankheitsverlauf zusammenhängen könnten, können durch klassische statistische Verfahren nicht erkannt werden“, erklärt Morrison. „Deshalb haben wir in unserer Konzeptstudie in silico, also computergestützt, virtuelle Patientinnen erstellt.“ Denn je größer die Datenmenge, desto besser können womögliche Muster erkannt werden, die für eine gute oder schlechte Prognose stehen.
Ein Generative Adversial Network (GAN, auf Deutsch: generierendes gegnerisches Netzwerk) ist ein Machine Learning Modell. Es besteht aus zwei gegnerischen neuronalen Netzwerken, die miteinander konkurrieren. Das eine Netzwerk – der Generator - erzeugt die Daten, das andere Netzwerk – der Diskriminator oder Prüfer - bestimmt, ob die Daten echt oder falsch sind. Die Daten, die so generiert werden, sind möglichst authentisch.
Um diese virtuellen Patientinnen zu erstellen, haben die beiden Wissenschaftler und Christoph Morhard von ProKanDo ein Modell aus dem Machine Learning als Vorbild angewandt, das zur Bildgenerierung verwendet wird, ein sogenanntes GAN Modell. „Der Deep Learning-Algorithmus lernte, wie er Bilder von genetischen Informationen erzeugen kann, die so täuschend echt sind, dass die Daten der virtuellen Patientinnen nicht mehr von den Daten der echten Brustkrebspatientinnen unterschieden werden konnten“, erzählt Cristiano Guttà, der diese Studie im Rahmen seiner Doktorarbeit durchführte. Das funktioniert so, dass die KI mit den Datensätzen der echten Patientinnen trainiert wurde und anschließend lernte, daraus noch weitere, künstliche Brustkrebsdaten zu erzeugen. „So konnte die KI wiederum aus diesem größeren Datensatz lernen und besser unterscheiden zwischen Patientinnen, die eine gute Prognose haben und Patientinnen, die eine schlechte Prognose haben“, ergänzt Guttà. Schlecht bedeutet in diesem Fall, dass der Krebs zurückkehrt oder fortschreitet und die Patientin stirbt, gut bedeutet, dass sie überlebt.
Mit der Methode der Bilderkennung Muster in genetischen Informationen erkennen
„Unser System mit der Bezeichnung T-GAN-D nutzt Deep-Learning Algorithmen, die ursprünglich für die Bildanalyse konzipiert wurden. Damit können wir Brustkrebspatientinnen mit hohem und niedrigem Risiko unterscheiden“, sagt Markus Morrison. Zu diesem Zweck werden die genetischen Informationen in Bilder umgewandelt, in denen jedes Pixel und auch die Werte dieser Pixel zueinander in ihrer räumlichen Anordnung die Ausgangsinformationen darstellen. Diese Bilder werden dann als Eingabe für einen Klassifikator verwendet, der nach dem Training die Patientinnen in die jeweilige Risikokategorie einteilt. „Die Ergebnisse zeigen, dass unser System etablierten prognostischen Markern in den untersuchten Datensätzen ebenbürtig ist“, so Morrison. „Sowohl die Umwandlung der genetischen Informationen in Bilder als auch die Verwendung der künstlichen Datensätze sind vielversprechende Ansätze, die in Zukunft ein Sprungbrett für individualisiertere Prognosen sein könnten, auch in Kombination mit klinisch etablierten prognostischen Markern.“
Ein Klassifikator ist ein Algorithmus, der verwendet wird, um Daten in verschiedene Kategorien einzuordnen. Er analysiert Merkmale der Datenmuster und trifft daraufhin Entscheidungen über die Zuordnung zu einer bestimmten Klasse oder Kategorie. Klassifikatoren werden häufig in der maschinellen Lern- und Datenanalyse eingesetzt, um Muster in Daten zu identifizieren und Vorhersagen zu treffen.
Doch bis jetzt ist es noch nicht so einfach möglich zu sagen, welche Gene genau dafür verantwortlich sind, ob die Prognose gut oder schlecht ist. Deshalb wollen die beiden Wissenschaftler weiter an dieser Methode forschen. „Zusammen mit klinischen Partnern wäre es zielführend, unser Verfahren weiter zu verbessern, zum Beispiel durch die Fokussierung auf die nach biomedizinischem Wissensstand wichtigsten Gene für den Krankheitsverlauf. Damit könnten wir testen, ob die Prognosen noch zuverlässiger würden“, sagt Cristiano Guttà. So könnten zukünftige Behandlungsstrategien schon früh gezielt angepasst werden.
Denn wenn man schon weiß, dass ein hohes Rückfallrisiko besteht, könnten zum Beispiel die Untersuchungstermine enger getaktet werden. Oder man könnte ergänzende Behandlungsmaßnahmen, die das Rückfallrisiko senken, die aber ohne eine sichere Prognose vielleicht nicht eingeleitet worden wären, bereits früher anwenden. Bei guten Prognosen dagegen könnte man den Patientinnen gegebenenfalls belastende Behandlungen ersparen.
Soll der Algorithmus weiter lernen?
„Zudem muss man sich überlegen, ob man den Algorithmus, der jetzt so trainiert ist, auf diesem Trainingsstand lässt oder erlaubt, dass er weiter lernt“, ergänzt Markus Morrison. Denn es gibt immer mehr und immer bessere Datensätze, die öffentlich zugänglich sind, und es kommen immer neue Erkrankte hinzu. Sobald der Algorithmus aber weiter lernt, ist das aus klinisch-diagnostischer Sicht schwierig, da die Tests für Diagnosen und Prognosen fixiert sind, das heißt, sie sind genau charakterisiert, sie haben eine gewisse Performance, die auch so hinterlegt ist, und sie werden überall identisch eingesetzt. „Aber in dem Fall hier könnte es dazu führen, dass das Modell sich die ganze Zeit weiterentwickelt. Und es ist für den normalen klinischen Alltag und die Zulassungsverfahren bisher nicht klar geregelt, wie man damit umgehen sollte“, so Morrison.
Ein weiteres Problem besteht darin, dass die Rohdaten international nicht das gleiche Format haben, weil zum Beispiel Labore unterschiedliche Methoden oder unterschiedliche Maschinen verwenden. Trotzdem freuen sich die beiden Wissenschaftler, dass sie mit ihrem trainierten GAN-Diskriminator (T-GAN-D) einen vielversprechenden Ansatz geschaffen haben, der für die verbesserte Prognostik bei Krebserkrankungen in Zukunft eine neue und wichtige Rolle spielen könnte.
Manuela Mild | SimTech Science Communication
Die Studie fand in Zusammenarbeit mit der Ludwigsburger Firma ProKanDo GmbH statt, die auf IT-Security und KI-Lösungen spezialisiert ist.
Zum Nachlesen
Guttà, Cristiano & Morhard, Christoph & Rehm, Markus. (2023). Applying a GAN-based classifier to improve transcriptome-based prognostication in breast cancer. PLoS computational biology. https://doi.org/10.1371/journal.pcbi.1011035
Über die Wissenschaftler

Cristiano Guttà stammt aus Italien, hat Biologie mit Schwerpunkt Zell- und Molekularbiologie studiert und seinen Master in einem Bioinformatik-Labor absolviert. Am Institut für Physik an der Universität in Bari hat er einen zweiten Master erworben, der die Entwicklung der digitalen Infrastruktur zum Inhalt hatte. So kam er zum Programmieren und in die IT und schließlich zu Markus Morrison an die Universität Stuttgart, wo er in einem SimTech-Projekt mit Fokus auf Datenanalyse und Künstliche Intelligenz promovierte. Jetzt forscht er am Institut für Zellbiologie und Immunologie.

Markus Morrison ist Professor und Leiter des Instituts für Zellbiologie und Immunologie an der Universität Stuttgart. Er hat Biologie mit einem Schwerpunkt in Biophysik studiert, interessierte sich dann für die Zellbiologie und arbeitete rund 15 Jahre in medizinischen Fakultäten, ein Großteil davon im Ausland. Seit er 2016 als Professor am Institut für Zellbiologie und Immunologie startete, beschäftigt er sich auch wieder mehr mit Grundlagenforschung. Translationale Forschung, also die Forschung, die klinisch relevant ist und eine klinische Perspektive hat, betreibt er weiterhin mit Kooperationspartnern und auch in einem Projekt in SimTech.